
SOLUTION ÉQUILIBRES EN
Dans de nombreux domaines de la chimie, les réactions ont lieu en solution. Citons les grandes préparations de la chimie inorganique, l'hydrométallurgie où l'on met en œuvre l'attaque des minerais par des solutions acides ou basiques, la biochimie, le nucléaire avec le retraitement des combustibles, l'analyse chimique avec les titrages volumétriques et coulométriques. On peut y adjoindre certaines réactions de la chimie organique.
La notion d' équilibre chimique tient une large place en chimie des solutions. Les réactions mises en jeu sont régies par les règles classiques découlant de la loi d'action de masses, pour autant qu'elles soient rapides. C'est en général le cas si on excepte bon nombre de réactions d'oxydoréduction et certaines réactions hétérogènes où il faut tenir compte de la vitesse de formation et d'évolution des précipités.
L'état d'une solution est en principe parfaitement défini par la connaissance des quantités introduites (les bilans-matière) et du modèle qui régissent l'ensemble des équilibres mis en jeu, chacun de ces équilibres se traduisant par une constante d'équilibre. Moyennant diverses simplifications, on peut obtenir des solutions approchées qui sont suffisantes pour certaines applications ; on peut maintenant, à l'aide des moyens de calcul modernes, résoudre tout problème de chimie des solutions de manière complète et rigoureuse.
L'introduction d'un soluté au sein d'un solvant implique l'existence d'interactions variées très énergétiques. L'interaction la plus simple tient son origine dans le caractère polaire des molécules de la plupart des solvants usuels – eau, alcools, amides... (interactions ion-dipôle ou dipôle-dipôle). Pendant la dissolution, le solvant exerce une action de solvatation (hydratation dans le cas de l'eau) au cours de laquelle chaque espèce dissoute (ion ou molécule) s'entoure d'un cortège de molécules de solvant. Il peut également exercer une action de solvolyse au cours de laquelle certaines liaisons sont rompues avec apparition d'espèces nouvelles. Le solvant constitue, en outre, un milieu diélectrique où les forces d'attraction s'exerçant entre ions de signes contraires sont affaiblies.
L'eau manifeste ces aptitudes de manière très marquée. Elle dissout un grand nombre de composés. Pour certains, les ions préexistant à l'état solide sont hydratés (cas du chlorure de sodium) ; pour d'autres, les liaisons sont ionisées (cas du chlorure d'hydrogène). La constante diélectrique très élevée (ε = 80) permet la séparation des ions hydratés (dissociation ionique), au point qu'ils peuvent être considérés comme indépendants.
Ces deux caractéristiques (pouvoir ionisant et pouvoir dissociant) de chaque solvant expliquent son rôle spécifique vis-à-vis des propriétés chimiques des espèces en solution.

Acides et bases
Planeta Actimedia S.A.© Encyclopædia Universalis France pour la version française.
Toutes les réactions en solution homogène se ramènent à trois types principaux : les réactions d'oxydoréduction, les réactions acide-base, les réactions de formation de complexes. Lorsqu'un composé a une solubilité limitée, il convient d'y ajouter les réactions de précipitation. Ces quatre types de réactions sont souvent mêlés. Des analogies entre les trois types de réactions en phase homogène apparaissent et, de ce fait, en adoptant la notion de couple donneur-accepteur d'une particule, un seul mode de raisonnement permet de les expliquer.
Réactions acide-base (transfert de protons)
Définitions
Acides et bases
Selon les concepts introduits par J. N. Brönsted, les acides sont des composés susceptibles de céder des protons : ce sont des donneurs de cette particule. Dans le même temps, les bases, composés à même d'en fixer, en sont des accepteurs. À tout acide correspond une base conjuguée[...]
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Claude COLIN : docteur-ès-sciences maître-assistant de première classe
- Alain JARDY : docteur-ès-sciences maître-assistant de première classe à l'Ecole Supérieure de Physique et de Chimie Industrielles de Paris
Classification
Pour citer cet article
Claude COLIN et Alain JARDY. SOLUTION ÉQUILIBRES EN [en ligne]. In Encyclopædia Universalis. Disponible sur : (consulté le )
Article mis en ligne le et modifié le 10/02/2009
Médias
Acides et bases
Planeta Actimedia S.A.© Encyclopædia Universalis France pour la version française.

Couples acide-base : constantes
Encyclopædia Universalis France

Répartition de l'acide o-phosphorique
Encyclopædia Universalis France
Autres références
-
ACIDES & BASES
- Écrit par Yves GAUTIER et Pierre SOUCHAY
- 12 364 mots
- 7 médias
 ...théorie de la dissociation ionique. Appliquée aux acides et bases, elle permit de préciser les notions précédentes et de les rendre quantitatives. Un acide HA est une substance qui, en solution aqueuse, fournit, lors de son équilibre de dissociation, des protons H+ :une base BOH est une substance...
...théorie de la dissociation ionique. Appliquée aux acides et bases, elle permit de préciser les notions précédentes et de les rendre quantitatives. Un acide HA est une substance qui, en solution aqueuse, fournit, lors de son équilibre de dissociation, des protons H+ :une base BOH est une substance...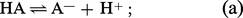
-
ACTIVITÉ, chimie
- Écrit par Dina SURDIN
- 260 mots
Grandeur introduite par G. N. Lewis, en 1907, pour exprimer les propriétés thermodynamiques des solutions. En effet, l'expression du potentiel chimique d'un composé dans une solution idéale, donné par la relation μ = kT ln N + μ0, où N représente la fraction molaire du composé,...
-
ÉLECTROCHIMIE
- Écrit par Jacques SIMONET
- 6 252 mots
- 10 médias
 ...des accepteurs (ou inversement) peut être prévue par la connaissance de grandeurs thermodynamiques (telles que ΔG, variation de l'enthalpie libre) des réactions équilibrées élémentaires vis-à-vis de l'électron :Cette réaction constitue un exemple de transfert d'électron homogène ensolution.
...des accepteurs (ou inversement) peut être prévue par la connaissance de grandeurs thermodynamiques (telles que ΔG, variation de l'enthalpie libre) des réactions équilibrées élémentaires vis-à-vis de l'électron :Cette réaction constitue un exemple de transfert d'électron homogène ensolution.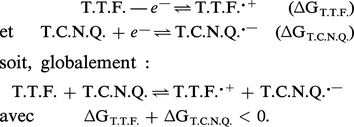
-
NITRIQUE ACIDE
- Écrit par Jean ROUXEL
- 1 604 mots
- 4 médias
 ...déjà 3,5 p. 100 des molécules HNO3. La dilution de l'acide pur dans de l'eau limite cet effet, déplace le dernier équilibre vers la gauche et provoque la réaction 3, de sorte que le bilan global s'identifie àl'équilibre classique d'un acide en solution dans l'eau (réaction 4).
...déjà 3,5 p. 100 des molécules HNO3. La dilution de l'acide pur dans de l'eau limite cet effet, déplace le dernier équilibre vers la gauche et provoque la réaction 3, de sorte que le bilan global s'identifie àl'équilibre classique d'un acide en solution dans l'eau (réaction 4).
