
STÉRÉOCHIMIE Stéréochimie organique
On trouvera dans l'article chimie organique le principe des méthodes qui ont conduit à l'établissement des formules développées planes. On va montrer ici pourquoi ces formules planes sont encore insuffisantes.
Désignant provisoirement par X, Y, Z, T (X′, Y′, Z′, T′) des substituants mono-atomiques tous différents, on est appelé à représenter par la formule plane (1) un dérivé tétrasubstitué du méthane, mais rien ne dit, a priori, si elle est équivalente à la formule (2).
L'expérience montrant qu'un tel dérivé n'existe que sous une seule forme, on serait tenté de penser que l'ordre de disposition des substituants autour d'un même atome est indifférent, et de couper court à toute discussion en écrivant le composé CX2YZ.
Mais, si l'on considère la formule à peine plus compliquée :
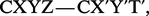
Cette discipline comprend deux parties : la stéréochimie statique, qui n'envisage que la structure spatiale des molécules, et la stéréochimie dynamique, qui étudie les variations des structures spatiales au cours des réactions.
Stéréochimie statique
Carbone « tétraédrique »
Tout composé CX4, CX3Y, CX2Y2, CX2YZ existe sous une seule variété. Il faut, ou bien admettre que si un tel composé peut, à l'origine, prendre naissance sous différentes formes, toutes se transforment spontanément en le composé unique le plus stable, ou bien que les composés obtenus par différentes voies sont, dès l'origine, identiques. En généralisant la première hypothèse, on serait amené à nier l'existence de plusieurs isomères pour CXYZ−CX′Y′T′, conclusion en opposition avec l'expérience. On a tenté de donner de la seconde hypothèse des démonstrations expérimentales ; elles ne sont pas à l'abri de toute critique, et il est plus raisonnable de la considérer comme un postulat qui se vérifiera par toutes ses conséquences.
L'unicité des composés CX4, CX3Y, CX2Y2, CX2YZ n'est compatible qu'avec une hypothèse structurale unique ; dans CX4, les quatre X jouent un rôle identique puisqu'il n'existe qu'un composé CX3Y, et tout couple YZ est interchangeable avec le couple ZY, puisqu'il n'existe qu'un composé CX2YZ. Il faut donc que les quatre substituants identiques dans CX4 occupent les quatre sommets d'un tétraèdre régulier dont le carbone occupe le centre. Dans les composés CX3Y, CX2Y2, CX2YZ, ce tétraèdre n'est plus régulier, mais l'édifice conserve au moins un plan de symétrie, montrant que les dispositions (3) et (4) sont absolument équivalentes.
Dans CX4, toutes les liaisons C−X ont même longueur, et les six angles de valence XĈX ont même valeur :

Dans les composés CX3Y, CX2Y2, CX2YZ, les distances interatomiques ne sont plus toutes égales et les angles de valence ne sont plus tous égaux ; toutefois, tant qu'il s'agit de molécules acycliques, ces angles s'écartent peu de 1090 28′.
Les structures réelles présentent les mêmes symétries qu'un tétraèdre régulier aux sommets duquel sont inscrites des lettres représentant les substituants. C'est pourquoi ce postulat est couramment désigné par « postulat du carbone tétraédrique ». En réalité, ni les faces ni les arêtes de ce tétraèdre n'ont un rôle quelconque ; seules les lignes de valence, axes de symétrie des orbitales, ont une signification physique et le postulat du carbone tétraédrique devrait s'énoncer : les axes des quatre orbitales du carbone ne sont pas dans un même plan, mais forment des angles égaux dans CX[...]
La suite de cet article est accessible aux abonnés
- Des contenus variés, complets et fiables
- Accessible sur tous les écrans
- Pas de publicité
Déjà abonné ? Se connecter
Écrit par
- Henri B. KAGAN : professeur de chimie à l'université de Paris-Sud, membre de l'Académie des sciences
- Charles PRÉVOST : professeur à la faculté des sciences de Paris, à l'École centrale des arts et manufactures de Paris et à l'École normale supérieure de Fontenay-aux-Roses
Classification
Pour citer cet article
Henri B. KAGAN et Charles PRÉVOST. STÉRÉOCHIMIE - Stéréochimie organique [en ligne]. In Encyclopædia Universalis. Disponible sur : (consulté le )
Article mis en ligne le et modifié le 14/03/2009
Médias

Réactions 63 à 83
Encyclopædia Universalis France

Réactions 45 à 53
Encyclopædia Universalis France

Réactions 11 à 26
Encyclopædia Universalis France
Autres références
-
AMPÈRE ANDRÉ-MARIE (1775-1836)
- Écrit par Louis POUDENSAN
- 1 788 mots
- 1 média
 ...s'arrête pas à l'énoncé de cette loi ; il cherche à en déduire la forme et l'arrangement des atomes pour prévoir leurs combinaisons, leurs substitutions. Il conçoit ce que l'on appelle aujourd'hui lastéréochimie qui, à son époque, ne fut considérée que comme « une fantaisie révolutionnaire ».
...s'arrête pas à l'énoncé de cette loi ; il cherche à en déduire la forme et l'arrangement des atomes pour prévoir leurs combinaisons, leurs substitutions. Il conçoit ce que l'on appelle aujourd'hui lastéréochimie qui, à son époque, ne fut considérée que comme « une fantaisie révolutionnaire ». -
BARTON DEREK HAROLD RICHARD (1918-1998)
- Écrit par Georges BRAM
- 484 mots
- 1 média
Chimiste britannique né à Gravesend (Kent). Derek Harold Richard Barton obtient son doctorat en 1942 à l'Imperial College de l'université de Londres, où il travaille deux ans dans un laboratoire dépendant des autorités militaires. Après un an passé dans l'industrie chimique, il retourne à l'Imperial...
-
CHIMIE THÉORIQUE
- Écrit par Lionel SALEM et François VOLATRON
- 4 288 mots
- 10 médias
 ...cas présent, de la symétrie spatiale des orbitales moléculaires. Woodward et Hoffmann ont considéré la réaction de fermeture de cycle de la molécule de butadiène. Ils ont fait remarquer que, pour une molécule portant des substituants différents X et Y aux deux bouts, deux produits distincts peuvent être...
...cas présent, de la symétrie spatiale des orbitales moléculaires. Woodward et Hoffmann ont considéré la réaction de fermeture de cycle de la molécule de butadiène. Ils ont fait remarquer que, pour une molécule portant des substituants différents X et Y aux deux bouts, deux produits distincts peuvent être... -
CHIRALITÉ, chimie
- Écrit par Pierre LASZLO
- 1 347 mots
Un objet est chiral s'il n'est pas superposable à son image dans un miroir, ou image spéculaire. Nos mains appartiennent à cette classe des objets chiraux, d'où leur nom, dérivé du grec kheir, « main ». Nos mains, gauche et droite, sont l'image spéculaire l'une de l'autre. De la...
- Afficher les 23 références
