

Maladies génétiques
Articles
-
ACATALASIE
- Écrit par Universalis
- 149 mots
L'acatalasie est une maladie héréditaire du métabolisme rare, causée par le déficit d'une enzyme, catalyseur organique, appelée catalase. Le déficit de l'activité de la catalase est présent dans de nombreux tissus de l'organisme, en particulier les globules rouges, la moelle osseuse, le foie et...
-
ALCAPTONURIE
- Écrit par Jacques HANOUNE
- 383 mots
Depuis 1822, on connaît l'alcaptonurie, une maladie très rare apparaissant dans l'enfance, associant une modification de la couleur de la peau, la présence d'urines foncées et une atteinte articulaire. En 1898, le médecin britannique Archibald Garrod identifiait la substance responsable...
-
ANDERSEN MALADIE D'
- Écrit par Universalis
- 193 mots
La maladie d'Andersen, également appelée glycogénose de type IV, est une maladie héréditaire du métabolisme très rare liée au déficit en amylo-1 :4,1 :6-transglucosidase, enzyme clé de la synthèse du glycogène. Ce déficit entraîne la production excessive d'une forme anormale de glycogène, l'amylopectine,...
-
ARNm THÉRAPEUTIQUES
- Écrit par Bruno PITARD
- 6 616 mots
- 5 médias

Un grand nombre de maladies, génétiques en particulier mais aussi acquises, pourraient être soignées si on pouvait introduire dans l’organisme, voire dans les cellules en cause, la ou les protéines capables de corriger l’anomalie à l’origine de la maladie. Face à ce besoin, on ne dispose que d'une...
-
CUTIS LAXA
- Écrit par Universalis
- 121 mots
Maladie héréditaire ou acquise rendant la peau anormalement élastique et plissée. Il s'agit d'une anomalie du tissu conjonctif élastique de la peau, dont l'élastine est le principal constituant. Il y a plusieurs formes de la maladie, que l'on classe en formes héréditaires et formes acquises. Les...
-
DALTONISME
- Écrit par Philippe LANTHONY
- 1 596 mots
- 2 médias
Le terme « daltonisme » désigne, dans le langage courant, une déficience de la vision des couleurs, ou dyschromatopsie. Le daltonisme est ainsi nommé en hommage au célèbre chimiste anglais John Dalton (1766-1844) qui le décrivit en 1794 en analysant sa propre vision colorée. Le daltonisme...
-
DÉFICIT EN GLUCOSE-6-PHOSPHATE DÉSHYDROGÉNASE
- Écrit par Universalis
- 234 mots
Maladie héréditaire du métabolisme caractérisée par une augmentation de la tendance à la destruction des globules rouges qui libèrent leur hémoglobine (hémolyse), en particulier après absorption de certains médicaments.
Comme son nom l'indique, la maladie est due à une diminution importante...
-
FABRY MALADIE DE
- Écrit par Universalis
- 187 mots
Également appelée angiokératome diffus, la maladie de Fabry est une maladie héréditaire liée au chromosome X qui est due au déficit de l'enzyme alpha-galactosidase A. L'absence de l'enzyme provoque l'apparition de dépôts anormaux de glycosphingolipides (céramide trihexoside) dans les vaisseaux...
-
FORBES MALADIE DE
- Écrit par Universalis
- 148 mots
La maladie de Forbes, également appelée maladie de Cori ou glycogénose de type III, est une maladie héréditaire rare dans laquelle la dégradation du glycogène en résidus de glucose simples est incomplète, entraînant l'accumulation de composés intermédiaires dans les cellules du ...
-
GAUCHER MALADIE DE
- Écrit par Universalis
- 289 mots
La maladie de Gaucher est une maladie héréditaire du métabolisme rare, caractérisée par une anémie, un retard mental et des anomalies neurologiques, une pigmentation jaunâtre de la peau et des lésions osseuses responsables de fractures pathologiques. La maladie de Gaucher est liée à...
Médias

Enzymopathies
Encyclopædia Universalis France

Hémoglobines et hémoglobinoses
Encyclopædia Universalis France

Hérédité daltonienne
Encyclopædia Universalis France

Ichtyose
Zhax/ Shutterstock
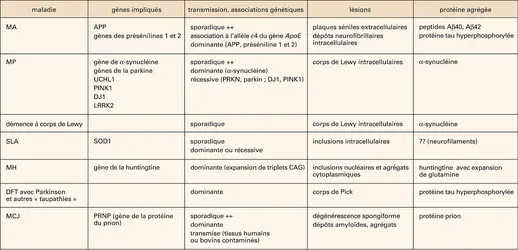
Maladies neurodégénératives
Encyclopædia Universalis France

Mécanisme d'accumulation des protéines anormales dans le tissu nerveux
Encyclopædia Universalis France

Microdélétions des autosomes
Encyclopædia Universalis France

Microduplications des autosomes
Encyclopædia Universalis France

Protéines impliquées dans les maladies neuromusculaires
Encyclopædia Universalis France

Téléthon 2007
P. Guibert/ AFM

Transmission génétique des hémoglobines anormales
Encyclopædia Universalis France
